一、背景
RNA-Seq实验设计中的“重复”包括:技术重复和生物学重复,重复是为了检测组间和组内的变异,对于假设检验至关重要。
- 技术重复为了估计测量技术(RNA-Seq)的变异。
- 生物学重复是为了发现生物组内的变异。简单的说,两组的基因表达的变化只有比组内变异还大时才能认为时显著的。
RNA-Seq试验中,抽样得到的raw read counts服从泊松分布。并且同一样本在两次试验中的结果不同,这称为shot noise。这种变异在RNA-Seq技术重复间成为Possion noise。生物学上不同的样本间的差异服从负二项(negative binomial)分布,有时称gamma-Poisson分布。由于RNA-Seq count数据也表现出zero inflation(大量值为0)的特征,所以很难拟合到负二项分布,所以有文章认为要用Poisson-Tweedie family建模。
基本方法就是以生物学意义的方式计算基因表达量,然后通过统计学分析表达量寻找具有统计学显著性差异的基因,从而
- 选择合适的基因
- 衡量结果的可靠性
分析方法
寻找差异表达基因有三种方式:
第一种是计算Fold change(倍数变化),十分简单粗暴的方法,计算方法如下:
- E = mean(group1) B=mean(group2)
- FC = (E-B) / min(E,B)
说人话就是,基因A和基因B的平均值之差与两者中较小的比值。选择2-3倍的基因作为结果(为什么是2-3倍,就是大家约定俗成)。
第二种就是统计检验,写文章的时候总需要给出一个p值告诉主编这个结果可信的(虽然p值也存在争论)。
复习一下:p值指的碰巧是拒绝零假设机会。P值越大假阳性越低,同时真实结果也可能会剔除。(注: 基因表达分析的零假设是: 基因在不同处理下的表达量相同。)
对于基因表达而言,研究目标是,对于同一个基因而言,他们之间的差异是处理不同造成,还是因为系统误差造成。
第三种:Fold Change + 统计检验。
在统计检验中你找到越多的差异表达基因,在p值矫正之后,你反而找不到差异表达基因。也就是说,如果在结果中存在大量滥竽充数的所谓的DE基因,那么在严格的p值矫正筛选后,反而会误删真实的DE基因。
因此在p值矫正之前,你先要手动剔除一部分明显就是假阳性的DE基因。这个步骤就需要用到前面的fold-change分析。
理论基础:线性模型, 设计矩阵和比较矩阵
线性回归 一般是用于量化的预测变量来预测量化的响应变量。比如说体重与身高的关系建模。基因表达可以简单写成, y = a + b · *treament* + e
方差分析(Analysis of Variance, ANAOVA)名字听起来好像是检验方差,但其实是为了判断样本之间的差异是否真实存在,为此需要证明不同处理内的方差显著性大于不同处理间的方差。
最简单的单因素方差分析,每一个结果都可以看成 yij = ai + u + eij, 其中u是总体均值,ai是每一个处理的差异,eij是随机误差。
上述两个公式非常相似,因为线性模型和方差分析都是广义线性模型(generalizing linear models, GLM)在正态分布的预测变量的特殊形式。
目前认为read count之间的差异是符合负二项分布,也叫gamma-Possion分布。那么问题来了,如何用GLM或者LM分析两个处理件的差异呢?其实可以简单的用上图的拟合直线的斜率来解释,如果不同处理之间存在差异,那么这个拟合线的斜率必定不为零,也就是与X轴平行。但是这是一种便于理解的方式(虽然你也未必能理解),实际更加复杂,考虑因素更多。
注1 负二向分布有两个参数,均值(mean)和离散值(dispersion). 离散值描述方差偏离均值的程度。泊松分布可以认为是负二向分布的离散值为1,也就是均值等于方差(mean=variance)的情况。
注2 这部分涉及大量的统计学知识,不懂就用维基百科一个个查清楚。
下面的设计矩阵(design matrix)就很好理解了, 其实就是用来告诉==不同的差异分析函数应该如何对待变量==。比如说我们要研究的==KD和control之间变化==,设计矩阵就是
| 样本 | 处理 |
|---|---|
| sample1 | control |
| sample2 | control |
| sample3 | KD |
| sample4 | KD |
那么比较矩阵(contrast matrix)就是告诉==差异分析函数应该如何对哪个因素进行比较, 这里就是比较不同处理下表达量的变化==。
二、RNA-seq的标准化
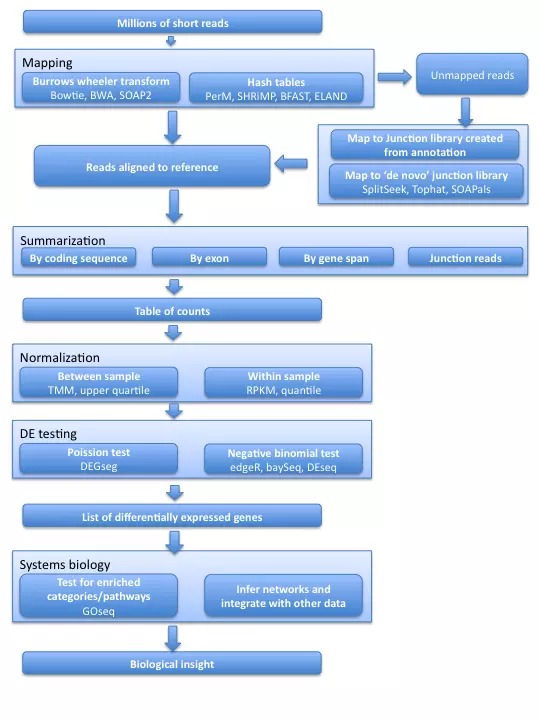
From RNA-seq reads to differential expression, Oshlack et al. Genome Biology 2010
基因表达标准化Normalisation
我们在比较不同样品不同基因的差异表达情况时,期望表达水平分布符合统计方法的基本假设,但由于测序深度和基因长度的不同,直接使用原始count分析会导致假阳性和假阴性过高,因此对原始数据进行标准化/均一化是十分必要的。
根据样本间和样本内重复可以把现有的诸多标准化方法大致分为两类,一类WSN(within-sample normalization):RPKM和quartile四分位数法;另一类BSN(between-sample normalization):TMM和upper quartile上四分位处理。
为了方便理解,假设目前你在一次测序中(即剔除批次效应)检测了一个物种的3个样本,A,B,C,这个物种有三个基因G1,G2,G3, 基因长度分别为100, 500, 1000. 通过前期数据预处理,你得到了尚未标准化的表达量矩阵,如下所示。
比较容易想到的标准化方法
基因表达量矩阵
| 基因/样本 | 样本A | 样本B | 样本C |
|---|---|---|---|
| G1(100) | 300 | 400 | 500 |
| G2(500) | 700 | 750 | 800 |
| G3(1000) | 1000 | 1300 | 1800 |
先说三个简单的策略,也就是最容易想到的方法
- Total Count, TC, 每个基因计数除以总比对数, 即文库大小, 然后乘以不同样本的总比对数的均值
- Upper Quartile, UQ, 和TC方法相似, 即用上四分位数替代总比对数
- Median, Med, 和TC方法相似, 用中位数代替总比对数
上面方法都相似,考虑到我的例子只有三个基因,所以只展示TC方法的结果. 可以发现,原本比其他组观测值低的A-G2,目前反而是最高(表格第二行)。
| TC处理后 | A | B | C |
|---|---|---|---|
| G1 | 370 | 402.7 | 418.1 |
| G2 | 863.3 | 744.1 | 543.5 |
| G3 | 1233.3 | 1308.8 | 1505.1 |
CPM (counts per million)
如果省去TC中的 “乘以不同样本的总比对数的均值” 这一步,那么差不多就是CPM (counts per million)的策略,也就是根据直接根据深度对每个样本单独进行标准化. 在edgeR和limma/voom里面都有出现过。
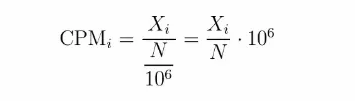
不同样品的测序量会有差异,最简单的标准化方式是计算counts per million (CPM),即原始reads count除以总reads数乘以1,000,000。
这种计算方式的缺点是容易受到极高表达且在不同样品中存在差异表达的基因的影响;这些基因的打开或关闭会影响到细胞中总的分子数目,可能导致这些基因标准化之后就不存在表达差异了,而原本没有差异的基因标准化之后却有差异了。RPKM、FPKM和TPM是CPM按照基因或转录本长度归一化后的表达,也会受到这一影响。
其它的标准化方法
为了解决这一问题,研究者提出了其它的标准化方法。
量化因子 (size factor, SF)是由DESeq提出的。其方法是首先计算每个基因在所有样品中表达的几何平均值。每个细胞的量化因子(size factor)是所有基因与其在所有样品中的表达值的几何平均值的比值的中位数。由于几何平均值的使用,只有在所有样品中表达都不为0的基因才能用来计算。这一方法又被称为 RLE (relative log expression)。
1 | calc_sf <- function (expr_mat, spikes=NULL){ |
上四分位数 (upperquartile, UQ)是样品中所有基因的表达除以处于上四分位数的基因的表达值。同时为了保证表达水平的相对稳定,计算得到的上四分位数值要除以所有样品中上四分位数值的中位数。
1 | calc_uq <- function (expr_mat, spikes=NULL){ |
TMM是M-值的加权截尾均值。选定一个样品为参照,其它样品中基因的表达相对于参照样品中对应基因表达倍数的log2值定义为M-值。随后去除M-值中最高和最低的30%,剩下的M值计算加权平均值。每一个非参照样品的基因表达值都乘以计算出的TMM。这个方法假设大部分基因的表达是没有差异的。
Median of Ratio (DESeq2)
该方法基于的假设是,即使处在不同条件下的不同个样本,大多数基因的表达是不存在差异的,实际存在差异的基因只占很小的部分那么我们只需要将这些稳定的部分找出来,作为标准化的内参,依据内参算出各个样本的标准化因子
(1)对每个基因计算几何平均数,得到一个假设的参考样本(pseudo-reference sample)
| gene | sampleA | sampleB | pseudo-reference sample |
|---|---|---|---|
| EF2A | 1489 | 906 | sqrt(1489 * 906) = 1161.5 |
| ABCD1 | 22 | 13 | sqrt(22 * 13) = 17.7 |
| … | … | … | … |
(2)对每个样本的每个基因对于参考样本计算Fold Change
| gene | sampleA | sampleB | pseudo-reference sample | ratio of sampleA/ref | ratio of sampleB/ref |
|---|---|---|---|---|---|
| EF2A | 1489 | 906 | 1161.5 | 1489/1161.5 = 1.28 | 906/1161.5 = 0.78 |
| ABCD1 | 22 | 13 | 16.9 | 22/16.9 = 1.30 | 13/16.9 = 0.77 |
| MEFV | 793 | 410 | 570.2 | 793/570.2 = 1.39 | 410/570.2 = 0.72 |
| BAG1 | 76 | 42 | 56.5 | 76/56.5 = 1.35 | 42/56.5 = 0.74 |
| MOV10 | 521 | 1196 | 883.7 | 521/883.7 = 0.590 | 1196/883.7 = 1.35 |
| … | … | … | … | … | … |
(3)获取每个样本中Fold Change的中位数,我们就得到了非DE基因代表的Fold Change,该基因就是我们选择的该样本的内参基因,它的Fold Change就是该样本的标准化因子
1 | normalization_factor_sampleA <- median(c(1.28, 1.3, 1.39, 1.35, 0.59))normalization_factor_sampleB <- median(c(0.78, 0.77, 0.72, 0.74, 1.35)) |
TMM (Trimmed Mean of M value, edgeR)
该方法的思想与DESeq2的Median of Ratio相同,假设前提都是:大多数基因的表达是不存在差异的
它与DESeq2的不同之处在于对内参的选择上:
- DESeq2选择一个内参基因,它的Ratio/Fold-Change就是标准化因子
- edgeR选择一组内参基因集合,它们对标准化因子的计算均有贡献:加权平均
TMM(trimmed mean of M value)方法出现在2010年,比TC、 UQ、Med, CPM方法高级一点,基本假设是绝大数的基因不是差异表达基因.计算方法有点复杂,简单的说就是移除一定百分比的数据后,计算平均值作为缩放因子,对样本进行标准化。这次我们用R/edgeR来算. 和之前不同,A组的G2基因标准化后还是最低,这就是trim所引起。
代表性软件edgeR,具体原理参见Footnotes[5]
1 | > library(edgeR) |
小结
DESeq2/DESeq有自己专门的计算缩放因子(scaling factor)的策略,它的基本假设就是绝大部分的基因表达在处理前后不会有显著性差异,表达量应该相似,据此计算每个基因在所有样本中的几何平均值(geometri mean), 每个样本的各个基因和对应的几何平均数的比值(fold change)的中位数就是缩放因子(scaling factor).
- 内参基因,House-keeping gene(s)。某一基因在样本中的表达肯定是一样的。(有限制,依赖基因的功能注释,数目少准确不高。)
- 通用方法:假设大多数基因是没有差异表达的。统计学方法找到标准化的因子。
- DESeq2(estimateSizeFactors/sizeFactors)、edgeR(TMM,calcNormFactors)。差异表达分析鉴定软件。
- 输入文件是reads count矩阵。
上述方法都是对样本整体进行标准化,标准化的结果只能比较不同样本之间的同一个基因的表达水平。
如果要同时比较不同样本不同基因之间的表达量差异,就得考虑到每个基因的转录本长度未必相同,毕竟转录本越长,打算成片段后被观察到的概率会高一点。此处不做展开。
对于差异表达分析而言,标准化不但要考虑测序深度的问题,还要考虑到某些表达量超高或者极显著差异表达的基因导致count的分布出现偏倚, 推荐用TMM, DESeq方法进行标准化。
三、DESeq2 分析差异表达
1. DESeq2差异分析基本流程
对于DESeq2需要输入的三个数据:表达矩阵、样品信息矩阵、差异比较矩阵
而对于DESeq2的差异分析步骤,总结起来就是三步:
- 构建一个dds(DESeqDataSet)的对象;
- 利用DESeq函数进行标准化处理;
- 用result函数来提取差异比较的结果。
2. 构建dds矩阵
- 构建dds矩阵的基本代码
1 | dds <- DESeqDataSetFromMatrix(countData = cts, colData = coldata, design= ~ batch + condition) |
- 其输入的三个文件:
- 表达矩阵:countData,就是通过之前的HTSeq-count生成的reads-count计算融合的矩阵。行为基因名,列为样品名,值为reads或fragment的整数。
- 样品信息矩阵:colData,它的类型是一个dataframe(数据框),第一列是样品名称,第二列是样品的处理情况(对照还是处理等)。可以从表达矩阵中导出或是自己单独建立。
- 差异比较矩阵:design,差异比较矩阵就是告诉差异分析函数哪些是对照,哪些是处理。
r代码
1 | raw_count <- read.csv('D:/1A/RNAseq/analysis/clone_dist.csv') |
Footnotes
- https://www.plob.org/article/11574.html
- https://mp.weixin.qq.com/s?__biz=MzI1MjU5MjMzNA==&mid=2247484514&idx=1&sn=1c6d227c6d7ac432d8baffb93b0b9072&chksm=e9e02dc3de97a4d59d918ee37655153fa4ccc8122e3259c0201ff441c922548f22f84311ad91&scene=21#wechat_redirect
- RNA-seq的标准化方法罗列
- 当我们在说RNA-seq reads count标准化时,其实在说什么?
- RNA-seq中的那些统计学问题
- DESeq2差异基因分析和批次效应移除-生信宝典.html